Our research focuses on developing computational and theoretical approaches to enable: materials synthesis prediction and rationalization; structure elucidation; and improved characterization across a diverse set of materials and applications. To do that, we often integrate different modeling tools, including high-performance computing (HPC), machine learning (ML), and atomistic simulations to model complex chemical phenomena and provide design principles to engineer different materials.
Some of our recent research projects include:
- developing algorithms and generative models to automate materials characterization
- creating computational methods to quantify information contents in atomistic simulations, and their relationship to ML and thermodynamics
- discovering synthesis recipes for materials using data-driven methods, theory, and simulations
- developing computational representations and algorithms applied to chemical systems
Atomistic machine learning
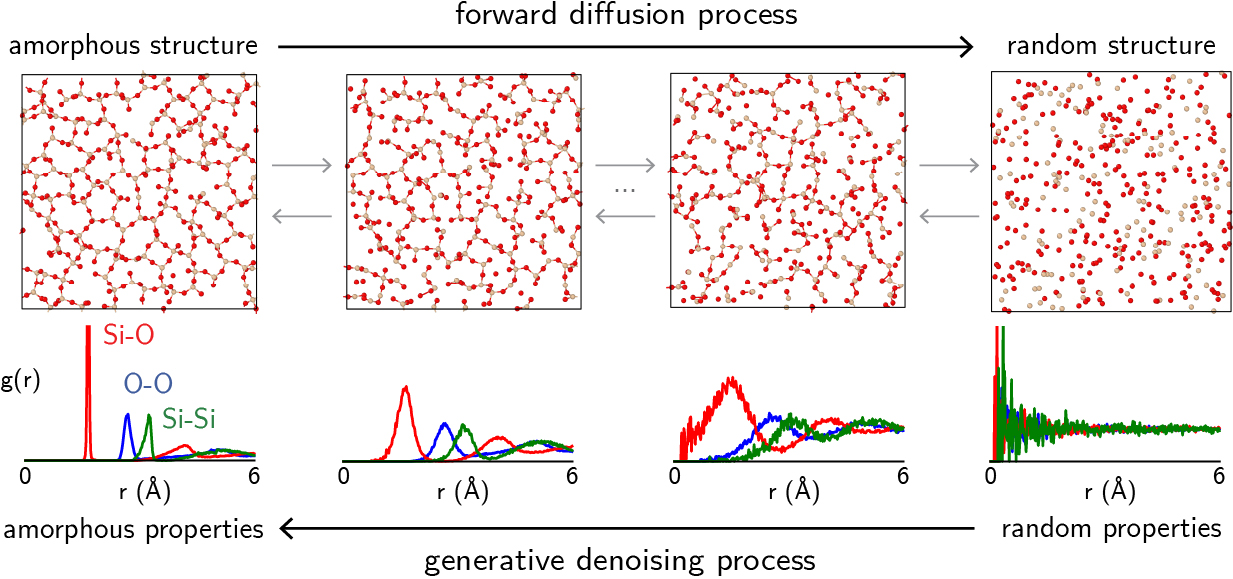
Artificial intelligence (AI) and machine learning (ML) are revolutionizing all areas of knowledge, including materials research. Particularly in the applied sciences, substantial efforts are devoted towards understanding robustness and uncertainty in materials simulations, as well as generative models for materials science.
For example, we recently proposed a generative model capable of generating amorphous materials with very high fidelity. The models were extensively validated by comparing them against structural features, macroscopic properties, and even experimental data, showcasing a reliable test suite for generative models. We also collaborated with colleagues at UCLA to understand how large language models can generate crystal structures without the need for fine-tuning.
Within ML interatomic potentials, we proposed new ways to improve the robustness of ML models for atomistic simulations by combining ideas on uncertainty quantification, chemical sampling, and adversarial attacks, as well as understanding why some ML models are more robust than others in production simulations. Recently, we have also proposed an information-theoretical perspective to explain trends in errors, detect outliers, and perform uncertainty quantification for machine learning potentials in a model-free approach. These information-theoretical methods also allow us to compress atomistic datasets with high efficiency compared to typical algorithms, showcasing how the combination of theory, simulations, and machine learning can be used to accelerate developments in materials modeling.
Literature-enabled materials synthesis
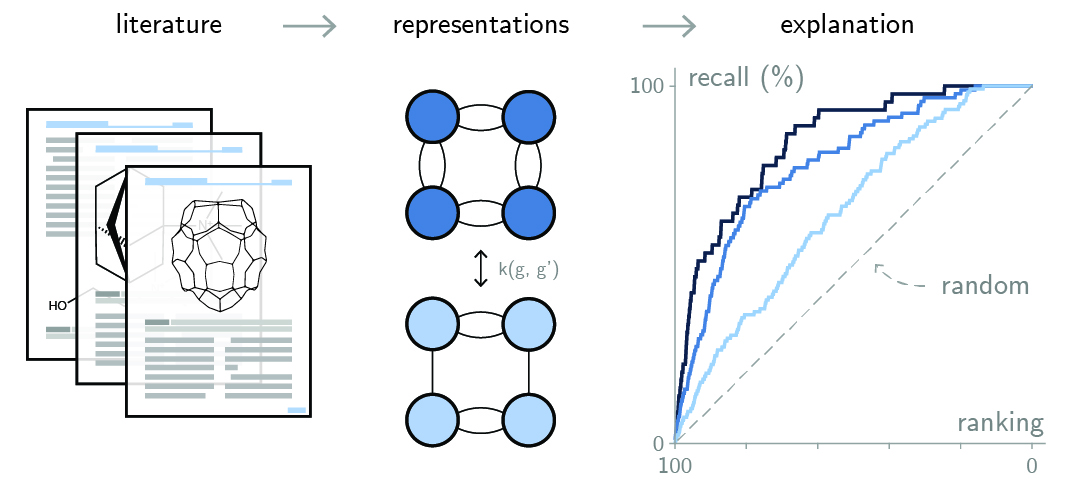
The scientific literature contains an immense amount of latent information, often accessible only through years of dedicated study. Automating the extraction of knowledge from this corpus of data can accelerate materials discovery, but correlating materials properties reported in the literature with a theoretical insight requires developing representations to interpret that data.
In our work, we combine representation learning to literature extraction to explain existing phenomena. For instance, we developed a graph-theoretical “order parameter” for explaining diffusionless transformations in zeolites that explains most of the cases of such polymorphic transformations in the literature. In another example, over six decades of literature are used to validate a theory on phase competition of nanoporous materials. As the information contained in papers is not used as training data for the model, it represents a ground truth dataset painstakingly curated by the field, and can be used for testing different hypotheses.
Automating and scaling up materials simulations

Automating data pipelines in the chemical sciences can expedite tasks such as simulations, inference, and data sharing. However, connecting different data provenances and deploying calculations to HPC centers requires substantial engineering work.
Our computational projects rely extensively on software and algorithm development for simulating and analyzing materials (see software page). We have developed computational platforms to facilitate the simulation of materials in distributed computing environments. In combination with its modular approach, our software provides great flexibility for simulating diverse materials systems.
Nanoporous materials design

The diversity of nanoporous materials compositions, structures, surfaces etc. provides wide potential for a variety of applications. In particular, the breadth of this chemical space poses a challenge when designing catalysts for given chemical reactions, especially when synthesis routes are taken into account. Our research provides theoretical insights to explore this structural and synthesis space using databases, simulations, and machine learning.
Examples of our work include exploring the synthesis of zeolites using high-throughput simulations, molecular descriptors, and machine learning which led to the discovery of new catalysts balancing lower synthesis costs, improved stability, and structural control. Recently, we had success in modeling inorganic synthesis conditions of zeolites, obtaining good agreement with patterns observed in the zeolite literature.
Our sponsors
Our group acknowledges the generous support of several organizations funding our research:







