Digital Synthesis Lab
(PI: Daniel Schwalbe-Koda)
We develop computational methods to enable predictive materials synthesis, thus accelerating their design beyond screening. Using a range of tools - from databases to machine learning - we propose solutions in energy, sustainability, and AI.
Recent Highlights
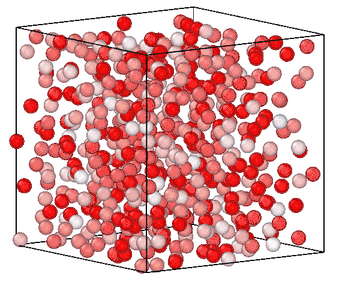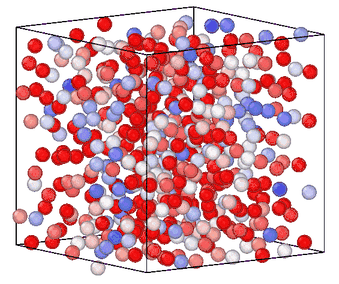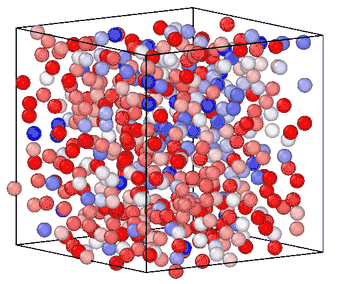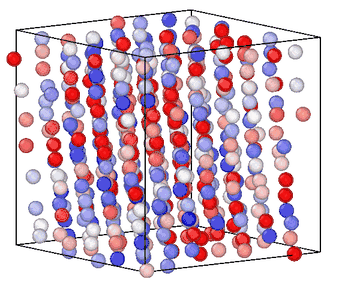
Information entropy is a general-purpose CV for enhanced sampling
Sampling rare events in molecular simulation often requires predefined knowledge of a collective variable (CV). We showed that information entropy of local atomic environments is a general-purpose CV for blind phase-space exploration. Our measure of entropy biases simulations toward low-probability, high-novelty configurations without predefined target states, system-specific order parameters or trained models. The method was demonstrated across five different systems spanning conformational sampling of organic molecules, homogeneous nucleation in copper, silicon glass formation, and graphite-to-diamond solid-state phase transformation, estalishing information entropy as a transferable tool for sampling and understanding rare events. [preprint]
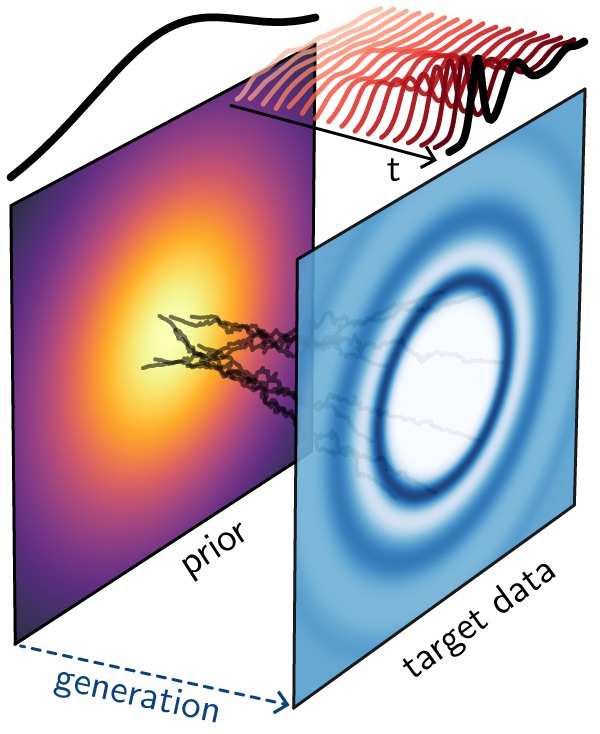
GLASS: Generative Learning of Amorphous Structures from Spectra
Elucidating structures of amorphous materials from spectroscopic data is a long-standing inverse problem limited by disorder, degeneracy, and reliance on expert oversight or expensive simulations. We developed GLASS, a generative framework that inverts multi-modal spectroscopic measurements into realistic structures without interatomic potentials or hand-crafted constraints. Systematic benchmarks across six spectroscopic modalities show that GLASS reliably generates out-of-distribution structures, with PDFs being particularly informative for models. We applied the framework to three contested experimental problems (paracrystallinity in amorphous silicon, a liquid–liquid phase transition in sulfur, and ball-milled amorphous ice), reproducing experimental spectra and revealing structural mechanisms inaccessible to diffraction analysis alone. This work can help automate the interpretation of spectroscopic data for amorphous structure elucidation at minimum computational cost. [preprint]

Automatic identification of compounds in mixtures from liquid-phase IR
Interpreting spectroscopy data is a critical bottleneck in automating chemical research and industrial characterization. Particularly within infrared (IR) spectroscopy, identifying compounds in complex, liquid-phase chemical mixtures largely relies on expert knowledge, as variable peak assignment, broadening, and shifts hinder data-driven methods. We showed that an algorithmic approach can identify components in both simulated and experimental mixture spectra with high accuracy despite nonlinearities in liquid-phase IR data. We applied the method to automatically interpret IR spectra in a large dataset of simulated liquid-phase IR, as well as experimental spectra, correctly identifying the components of nearly all samples within a blind study. This work provides tools and data to advance automated chemical laboratories through algorithmic interpretation of liquid-phase IR spectra of mixtures. [paper] [code] [data]
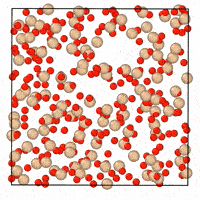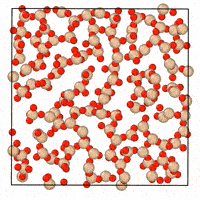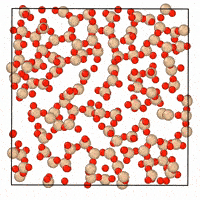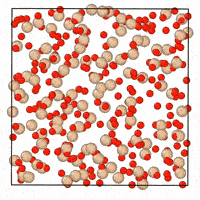
A generative diffusion model for amorphous materials
Generative models show ample promise for materials design, but face severe limitations in the amorphous materials space due to their complex structures. We developed a denoising diffusion framework that generates reliable atomistic structures across diverse amorphous systems and processing conditions while being up to three orders of magnitude faster than classical molecular dynamics simulations. Our model enables a range of applications in amorphous materials research, such as performing fracture simulations with large, slow-cooled structures, generating mesoporous structures, and augmenting experimental datasets with synthetic data. This work provides a roadmap on how to use, validate, and develop generative models for amorphous materials. [paper] [blog post] [data] [code]
Our main tools

Machine Learning

Simulation Workflows



