Digital Synthesis Lab
(PI: Daniel Schwalbe-Koda)
We develop computational methods to enable predictive materials synthesis, thus accelerating their design beyond screening. Using a range of tools - from databases to machine learning - we propose solutions in energy, sustainability, and AI.
Recent Highlights
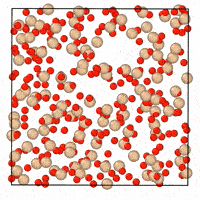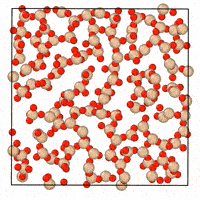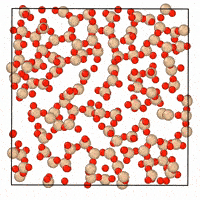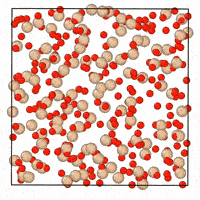
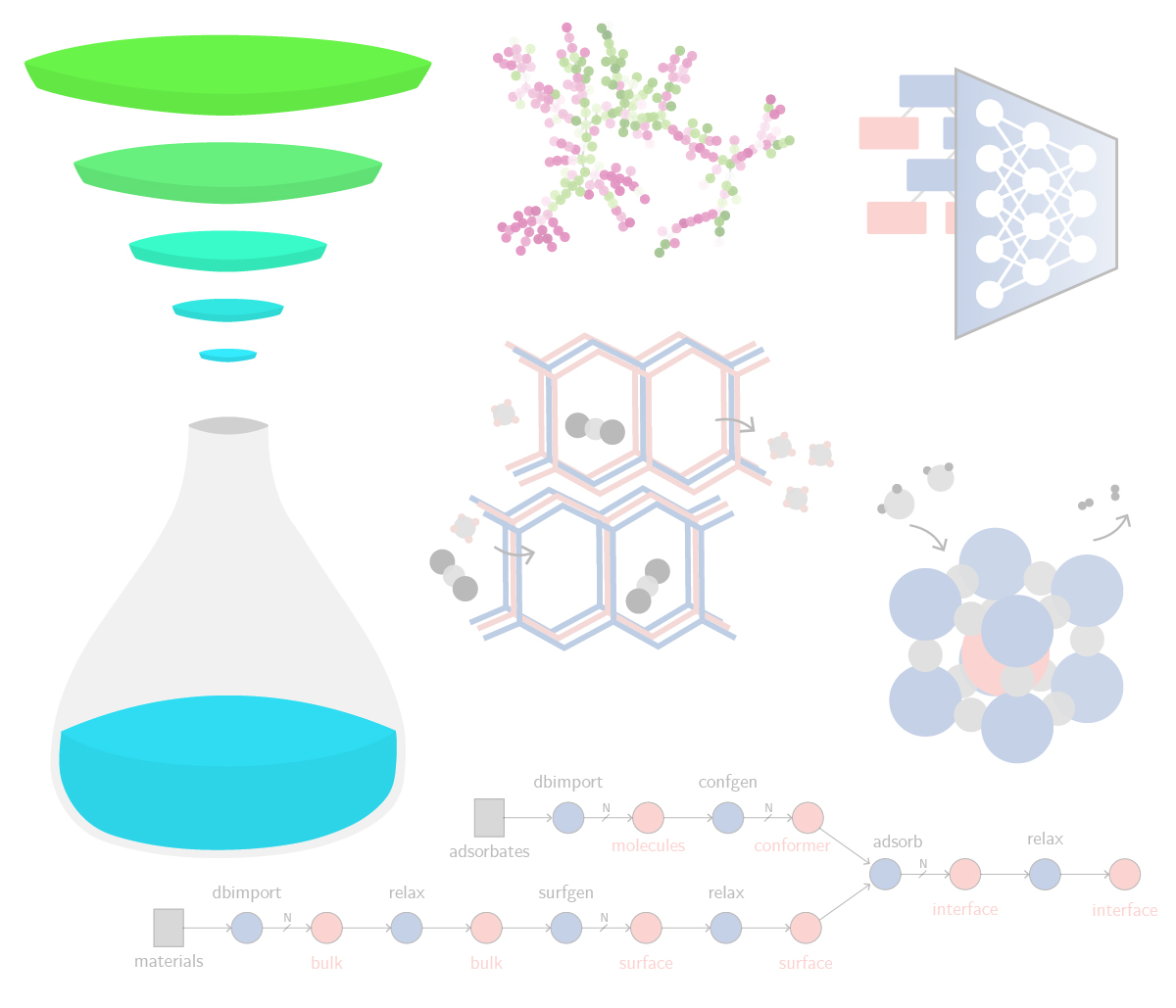
A generative diffusion model for amorphous materials
Generative models show ample promise for materials design, but face severe limitations in the amorphous materials space due to their complex structures. We developed a denoising diffusion framework that generates reliable atomistic structures across diverse amorphous systems and processing conditions while being up to three orders of magnitude faster than classical molecular dynamics simulations. Our model enables a range of applications in amorphous materials research, such as performing fracture simulations with large, slow-cooled structures, generating mesoporous structures, and augmenting experimental datasets with synthetic data. This work provides a roadmap on how to use, validate, and develop generative models for amorphous materials. [paper] [blog post] [data] [code]

Atomistic information theory for thermodynamics, UQ, and machine learning
Machine learning interatomic potentials (MLIPs) can bypass limitations of density functional theory (DFT) approaches regarding computational cost and scaling, but MLIPs often require heuristics, from training set selection to uncertainty quantification (UQ). By proposing an atomistic information theory, we showed that the information entropy from a distribution of local descriptors can be used in a range of problems in atomistic simulations, such as explaining trends in MLIP errors, rationalizing dataset analysis/compression, providing a robust UQ estimate for ML-driven simulations, and detecting outliers in atomistic simulations. We also proposed parallels between thermodynamic and information entropy and connect our information-theoretical approach to nucleation and growth. Our approach was demonstrated in a number of applications, and offers a general perspective on how materials theory, computation, and machine learning can be used to solve a range of problems in materials science. [paper] [code] [data]

Predicting coverage effects in catalysis using fast data pipelines
Modeling realistic interfaces using simulations is a challenge in computational catalysis. Especially in cases involving adsorbate-adsorbate interactions and co-adsorption, the number of configurations to evaluate grows rapidly with number of adsorbates, facet symmetry, and more, making DFT approaches overly expensive. On the other hand, machine learning potentials can be unreliable under generalization conditions and may require training pipelines relying on active learning. We proposed an efficient data generation strategy to control the extrapolation of neural network models and perform fast sampling in different coverage regimes. Our approach was demonstrated by sampling high-dimensional spaces of CO coverage on six copper facets, as well as the co-adsorption of CO and CHOH on Rh(111), showing good agreement with experimental results and revealing substantial differences in kinetic pathways depending on the coverage configuration. [paper] [code]
Our main tools

Machine Learning

Simulation Workflows



